Annotations and Queries
Annotations are key-value pairs stored as metadata for Projects, Files, Folders, and Tables that help users to find and query data. Annotations can be based on an existing ontology or controlled vocabulary, or can be created in an ad hoc manner and modified later as the metadata evolves. Annotations can be a powerful tool used to systematically group and/or describe things (files or data, etc.) in Synapse, which then provides a way for those things to be searched for and discovered.
For example, if you have uploaded a collection of alignment files in the BAM file format from an RNA-sequencing experiment, each representing a sample and experimental replicate, you can use annotations to add this information to each file in a structured way. Sometimes, users encode this information in file names, e.g., sampleA_conditionB.bam, which makes it “human-readable” but makes it difficult to search for in a systematic way, such as finding all replicates of sampleA_conditionB. Adding this information as Synapse annotations enables a more complete description of the contents of the File.
In this case, the annotations you may want to add might look like this:

Types of Annotations
Annotations can be one of four types:
- Text (Character Limit = 256)
- Integer
- Floating Point
- Date (Date and Time stored as a Timestamp)
How to Assign Annotations
Annotations may be added when initially uploading a file or at a later date. This can be done using the command line client, the Python client, the R client, or from the web. Using the programmatic clients facilitates batch and automated population of annotations across many files. The web client can be used to bulk update many files using file views.
Adding Annotations
To add annotations on a single entity through the web client, click the Annotations button in the upper right corner on a Project, Folder, or File page.
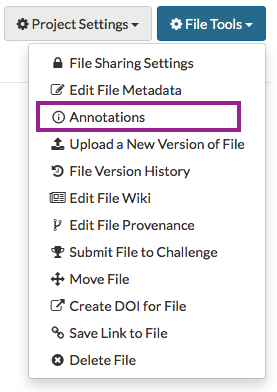
To add annotations on multiple files, please refer to our Synapse in Practice article Managing Custom Metadata at Scale for a tutorial on how to do this efficiently and effectively leveraging file views.
Command line
synapse store sampleA_conditionB.bam --parentId syn00123 --annotations '{"fileFormat":"bam", "assay":"rnaSeq"}'
Python
entity = File(path="sampleA_conditionB.bam",parent="syn00123")
entity.annotations = {"fileFormat":"bam", "assay":"rnaSeq"}
syn.store(entity)
R
entity <- File("sampleA_conditionB.bam", parent="syn00123")
entity <- synStore(entity, annotations=list(fileFormat = "bam", assay = "rnaSeq"))
Modifying Annotations
To update annotations on a single file:
Click File Tools, Annotations and Edit to add, delete, or modify annotations. Start by entering a key (the name of the annotation), select the type (text, integer etc.,), and enter the value. Click Save to store the annotations for this entity. To enter multiple Values for a single Key click Enter with the cursor in the Value field.
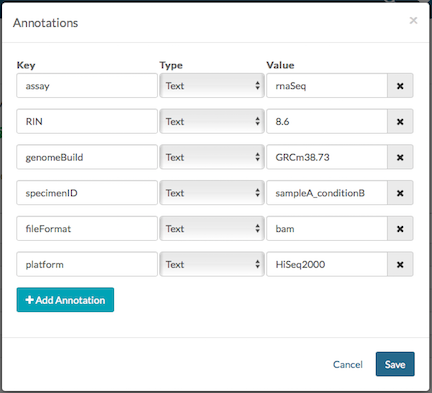
To add annotations on multiple files, please refer to our Synapse in Practice article Managing Custom Metadata at Scale for a tutorial on how to do this efficiently and effectively leveraging file views.
Command line
synapse set-annotations --id syn00123 --annotations '{"fileFormat":"bam", "assay":"rnaSeq"}'
Python
entity = syn.get("syn123")
# Please note that existing annotations will be overwritten. To modify ONLY one annotation:
entity.fileFormat = 'bam'
entity['fileFormat'] = 'bam'
#Please note that existing annotations will be overwritten
entity.annotations = {"fileFormat":"bam", "assay":"rnaSeq"}
syn.store(entity, forceVersion = F)
R
entity <- synGet("syn00123")
##### Modifying a set of annotations, preserving any existing annotations
existing_annots <- synGetAnnotations(entity)
synSetAnnotations(entity, annotations = c(existing_annots, list(fileType = "bam", assay = "rnaSeq")))
##### Add/update annotations, removing any other existing annotations
synSetAnnotations(entity, annotations = list(fileType = "bam", assay = "rnaSeq"))
Queries
Queries in Synapse look SQL-like and you can query any Table or View with <synId>.
SELECT * FROM <synId> WHERE <expression>
The
-
All entities (Projects, Files, Folders, Tables/Views, Docker containers):
id,name,createdOn,createdBy,modifiedOn,modifiedBy,etag,type,parentId,benefactorId,projectId -
Versionable entities (Files, Table/Views):
currentVersion -
Files only:
dataFileHandleId
Files also have an contentMd5, contentSize and contentType as properties. These properties are not available in a View and are not searchable.
SELECT * FROM syn123456 WHERE "id" = 'syn00012'
For a complete list of example queries, see:
Finding Files in a Specific Project
To find all files in a specific Project, create a View in the web client. For example, if you’d like to see all files in a Project, navigate to your project and then the Tables tab. From there, click Tables Tools and Add File View. Click Add container and Enter Synapse Id to create a tabluar file view that contains every file in the project, which you can now query. Importantly, if you want to later query on annotations, you must select Add All Annotations. For a more in-depth look at this feature, please read our articles on File Views.
Listing Files in a Specific Folder
To list the Files in a specific Folder, you need to know the synID of the Folder (for example syn1524884, which has data from TCGA related to melanoma). All entities in this Folder will have a parentId of syn1524884.
The function to find all Files in this Folder is called “getChildren”:
Python
foo = list(syn.getChildren(parent='syn1524884', includeTypes=['file']))
R
foo <- as.list(synGetChildren(parent='syn1524884', includeTypes=list('file')))
Queries on Annotations
If annotations have been added to Files, they can be used to discover files of interest from View syn123456.
For example, you can identify all Files annotated as bam files (fileFormat = bam) with the following query:
SELECT * FROM syn123456 WHERE "fileFormat"='bam'
Likewise, if you had put the RNA-Seq related files described in the section above into the project syn00123 with the described annotations, then you could find all of the files for conditionB and sampleA:
SELECT * FROM syn123456 WHERE "projectId"='syn00123' AND "specimenID"='sampleA_conditionB'
Lastly, you can query on a subset of entities that have a specific annotation. You can limit the annotations you want displayed as following.
SELECT specimenID,genomeBuild,fileFormat,platform FROM file WHERE "projectId"='syn00123' AND "specimenID"='sampleA_conditionB'
Reproducible queries can be constructed using one of the analytical clients (command line, Python, and R) and on the web client, query results can be displayed in a table on a wiki page.
In a project, from the wiki page click Wiki Tools in the upper right corner to Edit Project Wiki. Click Insert and choose Table: Query on Files/Folders. Enter your query in the box and click the Insert button. Once you save the wiki page, the results will be displayed as a table.
Command line
synapse query "SELECT specimenID,genomeBuild,fileFormat,platform FROM syn123456 WHERE \"specimenID\"='sampleA_conditionB'"
result = syn.tableQuery("SELECT specimenID,genomeBuild,fileFormat,platform FROM syn123456 WHERE \"specimenID\"='sampleA_conditionB'")
result = synTableQuery("SELECT specimenID,genomeBuild,fileFormat,platform FROM syn123456 WHERE \"specimenID\"='sampleA_conditionB'")
How to Download Based on Queries
You can download files in a folder using queries. Currently this feature is only available in the command line client. For example, if you want to download all files in a File View that has a synapse id of syn00123, use
synapse get -q "SELECT * FROM file WHERE parentId = 'syn00123'"
Troubleshooting
Single quotes in Synapse queries must be replaced by double quotes or two single quotes. In order to query for the chemicalStructure of 4'-chemical:
SELECT * FROM syn123 where "chemicalStructure" = '4"-chemical'
# OR
SELECT * FROM syn123 where "chemicalStructure" = '4''-chemical'
See Also
Need More Help?
Try posting a question to our Forum.
Let us know what was unclear or what has not been covered. Reader feedback is key to making the documentation better, so please let us know or open an issue in our Github repository (Sage-Bionetworks/synapseDocs).